Classes and functions related to genomics and proteomics. More...
Classes | |
| class | BamFile |
| class | InBamFile |
| class | OutBamFile |
| class | BamHeader |
| Wrapper around bam_hdr_t struct. More... | |
| class | BamPair |
| class | BamPairProxy |
| class | BamPairIterator |
| class | BamRead |
| Class holding a bam query. More... | |
| struct | BamLessPos |
| struct | BamLessEnd |
| struct | BamLessName |
| class | BamReadFilter |
| Filter bam reads. More... | |
| class | BamReadIterator |
| class to iterate through a InBamFile More... | |
| class | BamWriter |
| class | BamWriteIterator |
| Output iterator for bam file. More... | |
| class | Codon |
| struct | AminoAcidEqual |
| Functor comparing if two Codons translate to the same amino acid. More... | |
| class | DNA |
| struct | DnaComplementer |
| Functor that calculates genomic complement. More... | |
| class | Fasta |
| wrapper class around struct faidx_t in libhts More... | |
| class | GenomicPosition |
| class | GFF |
| class | GFF2 |
| class | GFF3 |
| class | Pileup |
Functions | |
| template<typename InputIterator , typename OutputIterator > | |
| void | dna_complement (InputIterator begin, InputIterator end, OutputIterator out) |
| template<typename BidirectionalIterator > | |
| void | dna_reverse_complement (BidirectionalIterator begin, BidirectionalIterator end) |
| template<typename BidirectionalIterator , typename OutputIterator > | |
| void | dna_reverse_complement_copy (BidirectionalIterator begin, BidirectionalIterator end, OutputIterator out) |
| const unsigned char | nt16_table (char) |
| const char | nt16_str (uint8_t) |
| template<typename RandomAccessIterator > | |
| double | alignment_score (const BamRead &bam, RandomAccessIterator ref, double mismatch, double gap=0.0, double open_gap=0.0) |
| unsigned short | chr2int (const std::string &str) |
| transform a string to unsigned short chromosome number More... | |
| double | phred (double p) |
| double | phred_inv (double x) |
Detailed Description
Classes and functions related to genomics and proteomics.
Function Documentation
| double theplu::yat::omic::alignment_score | ( | const BamRead & | bam, |
| RandomAccessIterator | ref, | ||
| double | mismatch, | ||
| double | gap = 0.0, |
||
| double | open_gap = 0.0 |
||
| ) |
Calculate alignment score based on bam's cigar and sequence and the reference as in [ref, ref + bam.end()-bam.pos()).
For indels the score is calculated as open_gap + len * gap. For mismatches the score is calculated -mismatch. For matches the score is unity.
For cigar element BAM_CMATCH the sequence and reference is used to differentiate match from mismatch.
Type Requirments:
RandomAccessIteratoris a Readable IteratorRandomAccessIteratoris a Random Access Traversal Iteratorvalue_typeis equality compareable withchar
- Since
- New in yat 0.13
| unsigned short theplu::yat::omic::chr2int | ( | const std::string & | str | ) |
transform a string to unsigned short chromosome number
If str starts with 'chr' that prefix is stripped away before translating the string to chromosome number. Function translates "X" to 23, "Y" to 24, and "M" or "MT" to 25. For other inputs utility::convert is used to transform the input to an unsigned short.
- Returns
- chromosome number
- Since
- New in yat 0.7
| void theplu::yat::omic::dna_complement | ( | InputIterator | begin, |
| InputIterator | end, | ||
| OutputIterator | out | ||
| ) |
bam_pair_analyse performs an operation on bam read pairs as defined by visitor. The function iterates over sorted input range of reads; if read is first read, it is cached for later use; if read is second read and mate is present in cache, visitor operates on pair, i.e., Visitor (mate, read) is called.
Type Requirements:
Iteratoris a Readable IteratorIteratoris a Single Pass IteratorIterator'sreferencetype must be convertible to BamReadVisitormust have anoperator()(BamRead, BamRead)(or anyconstor reference combination)
- Note
- Input range
[first, last) must be sorted or behaviour is undefined.
- Since
- New in yat 0.10 Function that transforms a sequence of chars to its genomic complement (see DnaComplementer). Sequence can be transformed in-place, i.e., begin and out can point to the same element.
Type Requirements:
InputIteratoris Readable IteratorInputIteratormodels Single Pass Iterator- value type of
InputIteratoris convertible to char OutputIteratormodels Incrementable IteratorOutputIteratoris a Writable Iteratorcharmust be convertible to value type of OutputIterator
- Since
- New in yat 0.14
| void theplu::yat::omic::dna_reverse_complement | ( | BidirectionalIterator | begin, |
| BidirectionalIterator | end | ||
| ) |
Function that transforms a sequence of chars to its genomic reverse complement, i.e., it transforms the sequence using DnaComplementer and reverse the sequence.
Type Requirements:
BidirectionalIteratoris Bidirectional IteratorBidirectionalIteratoris Writable Iterator- value type of
BidirectionalIteratoris convertible to char charmust be convertible to value type of BidirectionalIterator
- Since
- New in yat 0.14
| void theplu::yat::omic::dna_reverse_complement_copy | ( | BidirectionalIterator | begin, |
| BidirectionalIterator | end, | ||
| OutputIterator | out | ||
| ) |
Function that transforms a sequence of chars to its genomic reverse complement, i.e., it transforms the sequence using DnaComplementer and reverse the sequence.
Type Requirements:
BidirectionalIteratoris Readable IteratorBidirectionalIteratoris Bidirectional Traversal Iterator- value type of
BidirectionalIteratoris convertible to char charmust be convertible to value type of BidirectionalIterator
Type Requirements:
InputIteratoris Readable IteratorInputIteratormodels Single Pass Iterator- value type of
InputIteratoris convertible to char OutputIteratormodels Incrementable IteratorOutputIteratoris a Writable Iteratorcharmust be convertible to value type of OutputIterator
- See Also
- dna_complement
- dna_reverse_complement
- Note
- If input range and output range overlap, behaviour is undefined.
- Since
- New in yat 0.14
| const char theplu::yat::omic::nt16_str | ( | uint8_t | ) |
Convert a 4-bit encoded nucleotide to a character.
This is the same functionality that is provided in HTSLIB's global array seq_nt16_str, but this function works also when building against libbam. In libbam the array was called bam_nt16_rev_table.
This is the inverse of function nt16_table.
- Since
- New in yat 0.13
| const unsigned char theplu::yat::omic::nt16_table | ( | char | ) |
Convert nucleotide character to 4-bit encoding. The result is encoded as 1/2/4/8 for A/C/G/T or combinations of these bits.
This is the same functionality that is provided in HTSLIB's global array seq_nt16_table, but this function works also when building against libbam. In libbam the array was called bam_nt16_table.
This is the inverse of function nt16_str.
- Since
- New in yat 0.13
| double theplu::yat::omic::phred | ( | double | p | ) |
Phred scaled probability is defined as 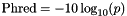
- Returns
- phred scaled probability
- Since
- New in yat 0.13
- See Also
- phred_inv
| double theplu::yat::omic::phred_inv | ( | double | x | ) |
The inverse of Phred scaling is calculated as 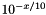
- Returns
- inverse of Phred scaling
- Since
- New in yat 0.13
- See Also
- phred
